


Role of NOD2 pathways genes in Sarcoidosis Cases with clinical characteristics of Blau syndrome
Bello et al. AJRCCM 2015;192:1133
Par Fleur Cohen Aubart, Service de médecine interne, Institut e3m, Pitié-Salpêtrière, Paris
Le syndrome de Blau est une maladie génétique autosomique dominante associée à des mutations dans le gène NOD2 et responsable d’un tableau de granulomatose débutant dans l’enfance avec des arthrites, une uvéite et des signes cutanés. NOD 2 est un récepteur intracellulaire intervenant dans l’immunité innée qui est capable d’interagir avec la protéine RIPK2 (voir l’image, issue de l’article), et activant les voies NFkappaB et la voie des MAP kinases. De façon intéressante, des polymorphismes de NOD 2 ont également été décrits comme facteur de risque de maladie de Crohn, une autre maladie granulomateuse.

Les auteurs ont cherché une association entre des polymorphismes (Single Nucleotide Polymorphism SNP) de 23 gènes de la voie NOD2 et la survenue d’une sarcoïdose (442 patients caucasiens et 1273 patients afro-américains). Ils n’ont pas observé d’association entre la survenue d’une sarcoïdose et des polymorphismes du gène NOD2 lui-même. En revanche, ils ont trouvé une association statistiquement significative avec un polymorphisme du gène MAPK13 (codant pour un des isoformes de la MAP kinase P38) chez les patients afro-antillais, et une association avec un haplotype du gène TAB2 surtout chez les patients caucasiens. Le complexe TAK1-TAB1-TAB2 est impliqué dans la cascade initiée par NOD2 et responsable d’une activation des macrophages, son inactivation induisant la mort des macrophages (voir schéma tiré de l’article).
Les auteurs ont également trouvé une association avec le gène TNFAIP3 codant pour une ubiquitine connue pour inhiber NFkappaB.
Commentaire du comité de lecture : cette étude suggère un rôle physiopathologique fort de la voie NOD 2/RIP/MAP kinases dans la formation des granulomes. L’existence de granulomes lors du syndrome de Blau associée à des mutations codant pour une protéine constitutionnellement active de NOD 2 était déjà un élément très intéressant pour expliquer la physiopathologie de la sarcoïdose. Ces résultats ouvrent la voie à de nouvelles pistes thérapeutiques qui pourraient cibler les MAP kinases ou les cytokines activées par cette voie.
Interferon-gamma-producing Th17.1 cells are increased in sarcoidosis and more prevalent than Th1 cells
Ramstein et al, AJRCCM publié le 9 decembre 2015 : 10,1164/rccm,201507-1499OC
Par Raphael Lhote et Fleur Cohen Aubart, Service de médecine interne, Institut e3m, Pitié-Salpêtrière, Makoto Miyara et Guy Gorochov, Département d’immunochimie et INSEM CERVI U1135
La sarcoïdose est habituellement décrite comme une maladie à polarisation Th1 du fait de l’infiltration tissulaire par des lymphocytes T CD4+ producteurs d’interféron-γ. Cependant, les cellules Th17 peuvent ressembler à des cellules Th1 et l’hypothèse des auteurs est que la voie Th17 pourrait être impliquée dans la physio pathogénie de la sarcoïdose, les cellules Th17 ayant été « confondues » avec des cellules Th1. Une étude antérieure rapportait notamment la présence de cellules dans le LBA de patients avec une sarcoïdose.
Les auteurs ont donc cherché à caractériser avec des méthodes immunologiques récentes les populations lymphocytaires, en particulier T CD4+, dans le LBA et le sang de patients ayant une sarcoïdose. La méthodologie employée était robuste avec une cohorte de patients américains principalement caucasiens comparée à des témoins dits « sains » dont l’âge était en moyenne inférieur puis une cohorte européenne de réplication où des patients principalement caucasiens à nouveau étaient comparés à des patients ayant une pneumopathie communautaire ou une BPCO.
Les éléments majeurs apportés par cette étude étaient que :
- on trouve dans le LBA des patients ayant une sarcoïdose essentiellement des cellules T CD4+ effectrices mémoires et peu ou pas de cellules T CD4+ naïves. Cette constatation conforte l’hypothèse précédemment émise que la sarcoïdose est une maladie déclenchée par une stimulation antigénique puisqu’il avait déjà été retrouvé dans le sang et le poumon de patients ayant une sarcoïdose une expansion oligoclonale de cellules T exprimant un récepteur αβ.
- Parmi les cellules T CD4+ effectrices mémoires trouvées dans le sang et le poumon des patients ayant une sarcoïdose, une forte proportion (environ 50 %) exprime le récepteur CCR6, un marqueur (bien que non totalement spécifique) des cellules Th17. Ces cellules CCR6+ ont une capacité à produire après stimulation pour la plupart de l’IFN γ seul, et pour une plus faible proportion à la fois de l’IFN γ et de l’IL17A ou l’IL17A seule. Il existerait donc 3 populations de Th17 chez les patients ayant une sarcoïdose.
- Des marquages des cellules T capables de produire l’IFN γ seul avec le récepteur CXCR3 (présent sur les Th17 mais pas les Th1) en plus de CCR6 (marqueurs plutôt des Th17) et CCR4 (marqueur de cellules Th2) ont permis de discriminer 3 populations de cellules T produisant de l’IFN γ impliquées dans la sarcoïdose : Th1 (produisant de l’IFN γ mais pas d’IL 17), Th17 (produisant de l’IL17 mais peu d’IFN γ) et Th17.1 (produisant de l’IFN γ et peu d’IL 17).
Au final, les auteurs concluent que la sarcoïdose, initialement caractérisée comme une maladie Th1, pourrait être plutôt caractérisée par une expansion de cellules Th17.1, cellules CD4+ produisant certes de l’IFN γ, mais également un peu d’IL 17A. Ces cellules constituent jusqu’à 60 % des cellules CD4+ des LBA de patients ayant une sarcoïdose. Ces résultats ont été répliqués dans la cohorte européenne.
Commentaire du groupe de lecture : cette étude a appliqué une méthodologie robuste et des méthodes immunologiques indiscutables qui ont permis de mieux caractériser la population T CD4+ trouvée dans le poumon de patients ayant une sarcoïdose. Les résultats ne modifient pas fondamentalement notre vision de l’orientation immuno-pathologique de cette affection, puisque les auteurs confirment que les effecteurs lymphocytaires sont malgré tout principalement sécréteurs d’IFN γ, et donc à orientation Th1 dominante. Toutefois, la question d’un intérêt d’anticorps monoclonaux ciblant l’IL 17A reste ouverte. Dans une étude sur la maladie de Crohn dans laquelle l’IL17A joue un rôle pro-inflammatoire probable (Ramesh, J Exp Med 2014), le secukinumab (anticorps monoclonal humanisé anti-IL 17A) entraîne des poussées de la maladie. Des études ultérieures précisant les rôles des sous-populations Th17 et leurs régulateurs (l’IL23 notamment) apporteront probablement des avancées supplémentaires.
High-density genetic mapping identifies new susceptibility variants in sarcoidosis phenotypes and shows genomic-driven phenotypic differences.
Rivera NV, et al. AJRCCM 2015 Dec 10. [Epub ahead of print]
Par François-Xavier Danlos et Fleur Cohen Aubart, Service de médecine interne, Institut e3m, Pitié-Salpêtrière
Dans cette étude internationale, les auteurs ont recherchés et analysés les polymorphismes génétiques / Single Nucleotide Polymorphismes (SNP) présents chez des patients atteint de sarcoïdose, en comparant trois populations : syndrome de Löfgren (LS), sarcoïdoses non Löfgren (non-LS) et une population témoin.
Ils étudient 4 cohortes de malades (7766 sujets, 1989 atteints de sarcoïdose). Trois sont européennes : Allemande (64LS, 413 non-LS, 4498 témoins), Hollandaise (241LS, 180 non-LS, 192 témoins), Tchèque (47LS, 263non-LS, 211 témoins) ; et une des Etats-Unis avec une population exclusivement dite Afro-américaine (781non-LS, 876 témoins).
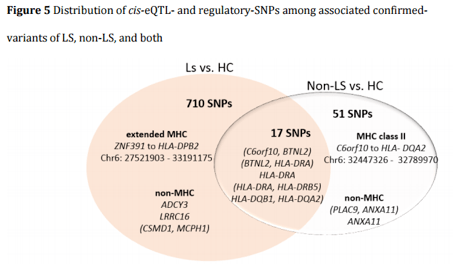
Parmi les patients atteints d’un LS les auteurs ont identifié une association avec les SNP rs3130288 en 3U’-UTR du gène ATF6B (ou CREBL1) localisé dans la région du CMH de classe III, et des SNP localisés au sein des régions codantes pour les CMH de classe I et II (LRRC16 et HLA-DPB2). En dehors des régions des CMH ils ont identifiés le SNP rs13407913 localisé dans un intron du gène ADCY3 du chromosome 2. Par ailleurs, en stratifiant les analyses en fonction du statut HLA-DRB1*03 (connu associé au LS), ils identifient chez les porteurs le SNP rs2248462, situés dans l’inter région des gènes HCP5 et MICB, et les non-porteurs, le SNP rs3104407 situés entre les gènes HLA-DQB1 et HLA-DQA2.
Parmi ceux atteints d’une sarcoïdose non-LS, le SNP principal est rs1964995 situé entre les gènes codant pour HLA-DR et HLA-DRB5 (CMH de classe II). Une cinquantaine de SNP ont été identifiés dans les régions situées entre les gènes C6orf10 et HLA-DQ2 sur le chromosome 6, et entre PLAC9 et ANXA11 sur le chromosome 10. Considérant le groupe non-LS très hétérogène en terme de phénotype clinique, ils ont comparés ceux atteint d’une maladie à manifestations extra-respiratoire (451 sujets) aux sujets sains, et identifient de nouveau le SNP rs1964995 comme étant celui le plus associé.
Par ailleurs, en effectuant une autre analyse des variants génétiques entre les trois populations, ils identifient 179 loci associés aux LS (de SCGN à HLA-DPB1 localisés dans les régions du CMH sur le chromosome 6) et 5 loci associés aux non-LS (BTNL2, HLA-DRA, HLA-DRB1 sur le chromosome 6 et PLAC9 et ANXA 11 sur le chromosome 10). Ces gènes sont superposables à ceux identifiés par immunochip.
L’analyse « fonctionnelle » des SNP identifiés montre qu’ils pourraient participer aux voies de signalisation permettant la réponse immune, la régulation, et la différenciation des lymphocytes T. Ainsi, il est intéressant de noter que certains polymorphismes identifiés sont « spécifiques » d’un phénotype de la maladie, d’autres sont conjoints aux formes LS et non-LS (figure 5, tirée de l’article).
Cette étude compare pour la première fois l’architecture génétique de différentes formes cliniques de sarcoïdose. Ainsi, 175 loci sont associés aux LS et 11 loci aux non-LS dans des cohortes indépendantes. Cinq sont communs aux deux phénotypes. Ces analyses permettent de justifier que les sarcoïdoses dites « LS et non-LS » ont une susceptibilité génétique différente et peut être des mécanismes immunitaires différents.
Commentaire du comité de lecture : Cette analyse intégrée ayant inclus des analyses génétiques, de transcription de et voies de signalisation indiquent que les phénotypes de sarcoïdose sont drivés par une susceptibilité génétique et des distributions génomiques particulières. La force de cette étude est l’utilisation de cohortes variées avec réplication des effets. Des explorations futures sont nécessaires pour mieux caractériser les régions dans lesquelles les SNP d’intérêt ont été trouvées.