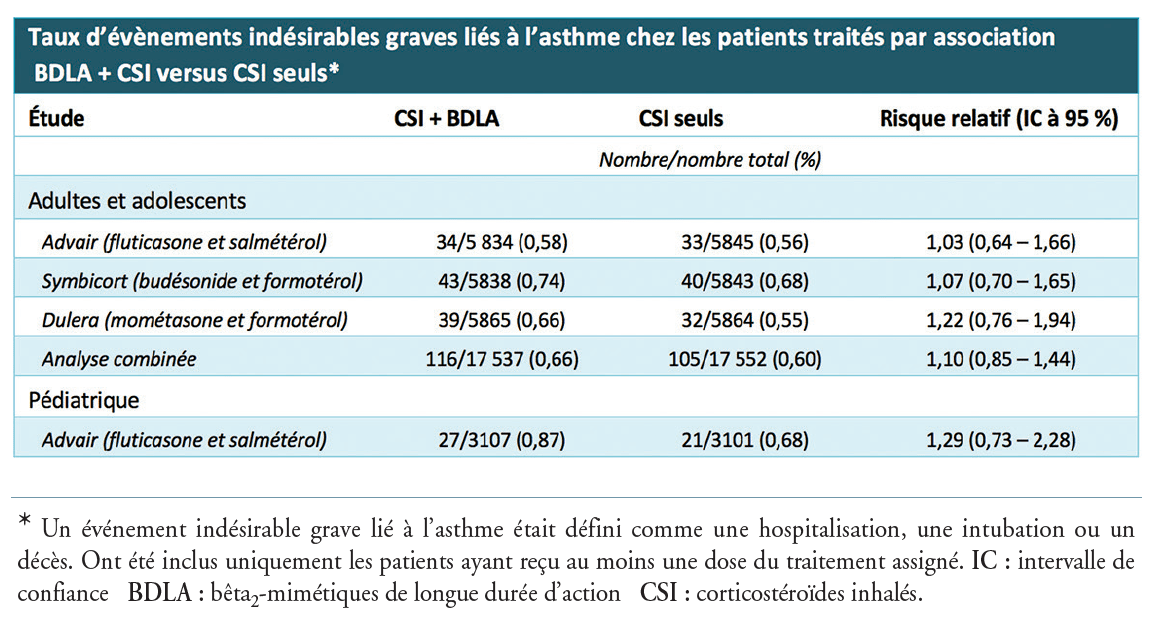
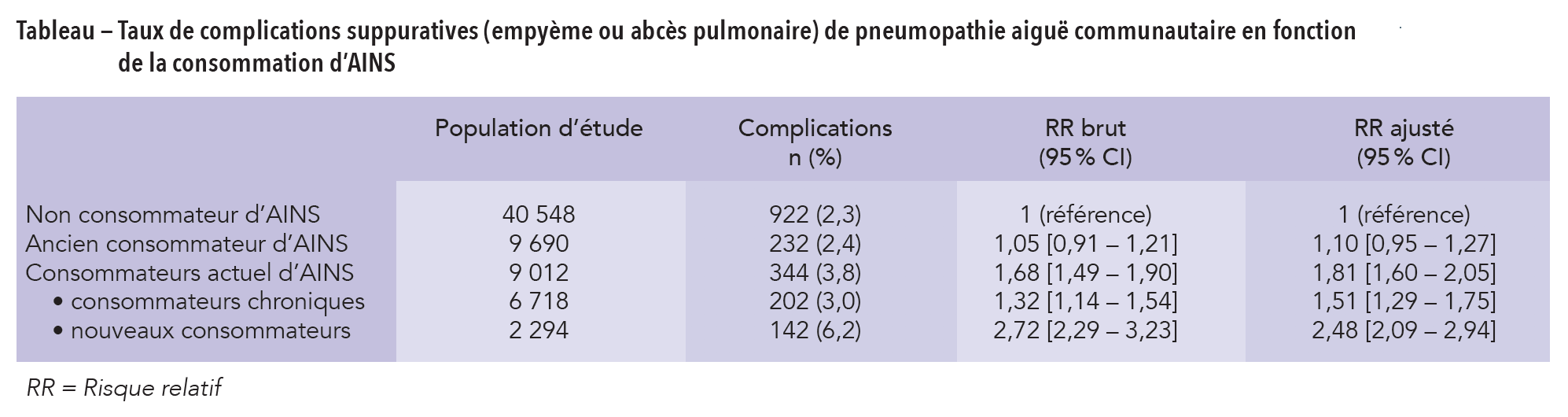
Fluoroquinolones par voie systémique ou inhalée : en plus du risque accru de tendinites, voici celui des anévrismes et de dissection aortique
Une information de plus à faire connaître aux patients. En octobre dernier, les laboratoires commercialisant des médicaments à base de fluoroquinolones, l’Agence européenne des médicaments (EMA) et l’Agence nationale de sécurité du médicament et des produits de santé (ANSM) ont émis une mise en garde concernant le risque de survenue d’anévrisme et de dissection aortiques associé à l’utilisation des fluoroquinolones par voie systémique ou inhalée.1
Sont ainsi en cause la ciprofloxacine, la lévofloxacine, la moxifloxacine, la norfloxacine, la fluméquine et l’ofloxacine. Les études épidémiologiques1 3 4rapportent un risque environ deux fois plus élevé de survenue d’anévrisme et de dissection aortique chez les patients prenant 5.] des fluoroquinolones par voie systémique, en comparaison avec les patients ne prenant pas d’antibiotiques ou prenant d’autres antibiotiques (amoxicilline) ; les personnes âgées présentent un risque encore plus élevé. Une étude non clinique 6 a montré que la ciprofloxacine augmente la susceptibilité à la dissection et à la rupture aortique dans un modèle de souris. Ce résultat est probablement en relation avec un effet de classe des fluoroquinolones similaire à celui impactant les tissus tendineux conduisant à un risque majoré de troubles tendineux.
Chez les patients présentant un risque de survenue d’anévrisme et de dissection aortique, les fluoroquinolones ne doivent être utilisées qu’après une évaluation attentive du rapport bénéfice/risque et après prise en compte des alternatives thérapeutiques. Les facteurs prédisposant à la survenue d’un anévrisme et d’une dissection aortique comprennent les antécédents familiaux d’anévrisme, la préexistence d’un anévrisme ou d’une dissection aortique, le syndrome de Marfan, le syndrome vasculaire d’Ehlers-Danlos, l’artérite de Takayasu, l’artérite à cellules géantes (ou maladie de Horton),la maladie de Behçet, l’hypertension artérielle et l’athérosclérose.
En pratique, les patients doivent être informés du risque d’anévrisme et de dissection aortique. Ils doivent être avertis de la nécessité d’une prise en charge immédiate par un médecin au sein d’un service d’urgence en cas d’apparition brutale d’une douleur intense abdominale, thoracique ou dorsale.
[hr]
Nicolas Postel-Vinay, Hôpital Européen Georges-Pompidou
L’auteur n’a déclaré aucun lien d’intérêt en relation avec cet article.
Communiqué de l’ANSM octobre 2018
InfoRespiration N°148 – Décembre 2018